Computational method development on CRISPR base editor screen
Aug 2021-
CRISPR base editors have opened a way to investigate the effect of individual variant to the phenotype. In SpRY-base editor screens, virtually all variants can be edited and by selecting cells based on the phenotype we can screen the variants that have effect to the phenotype of interest.
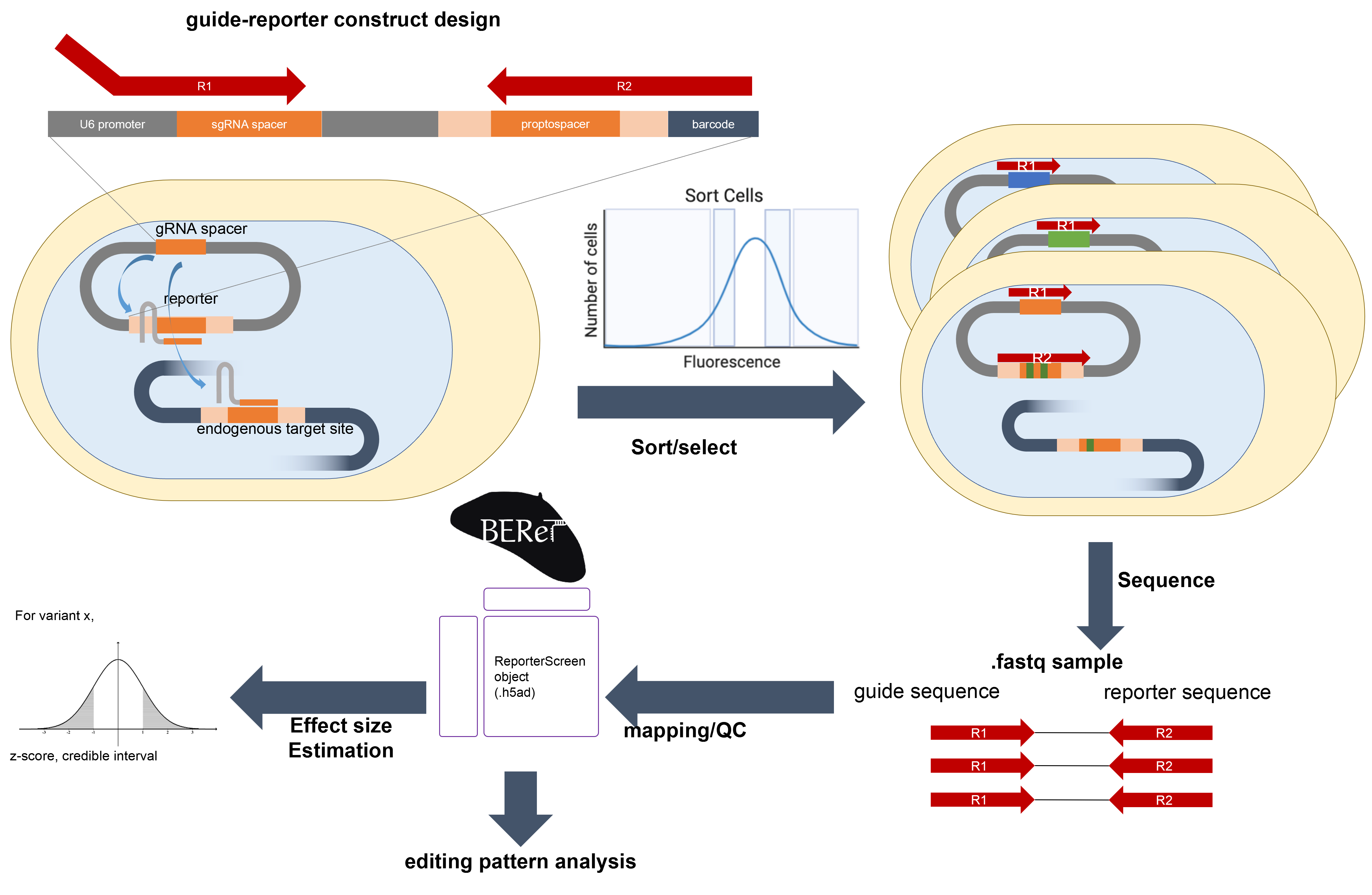

As part of the NIH IGVF consortium and in collaboration with Sherwood lab, I am developing computational method to better identify the functional variants in CRISPR base editor screens. Specifically, I'm working on developing the screen analysis method that takes the variable editing efficiency and pattern into account that utilizes stochastic variational inference (SVI) through probabilistic programming language. The model directly recapitulates count data generation process which provides direct interpretability.
SIMBA, a graph representation learning based single cell embedding method
Jan 2021-
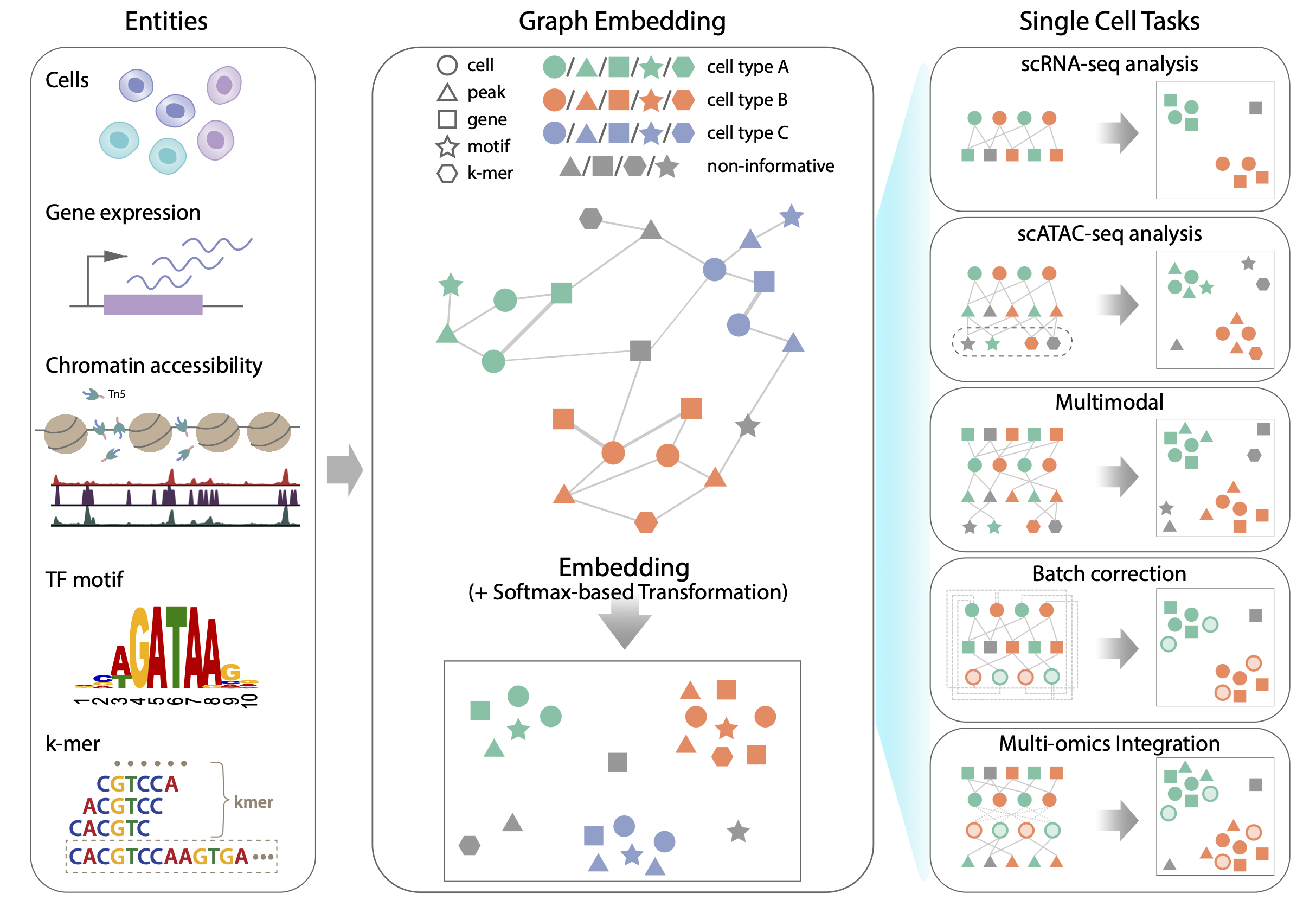 Our team developed SIMBA, a single cell embedding method that utilizes PyTorch-BigGraph, a multirelational graph embedding method in collaboration with Facebook Research.
Our team developed SIMBA, a single cell embedding method that utilizes PyTorch-BigGraph, a multirelational graph embedding method in collaboration with Facebook Research.
By formulating the single cell genomics data as the graph with cell and feature nodes, we are able to simultaneously learn the relationship between cells and multiple features, and the regulatory relationship between the different type of features.
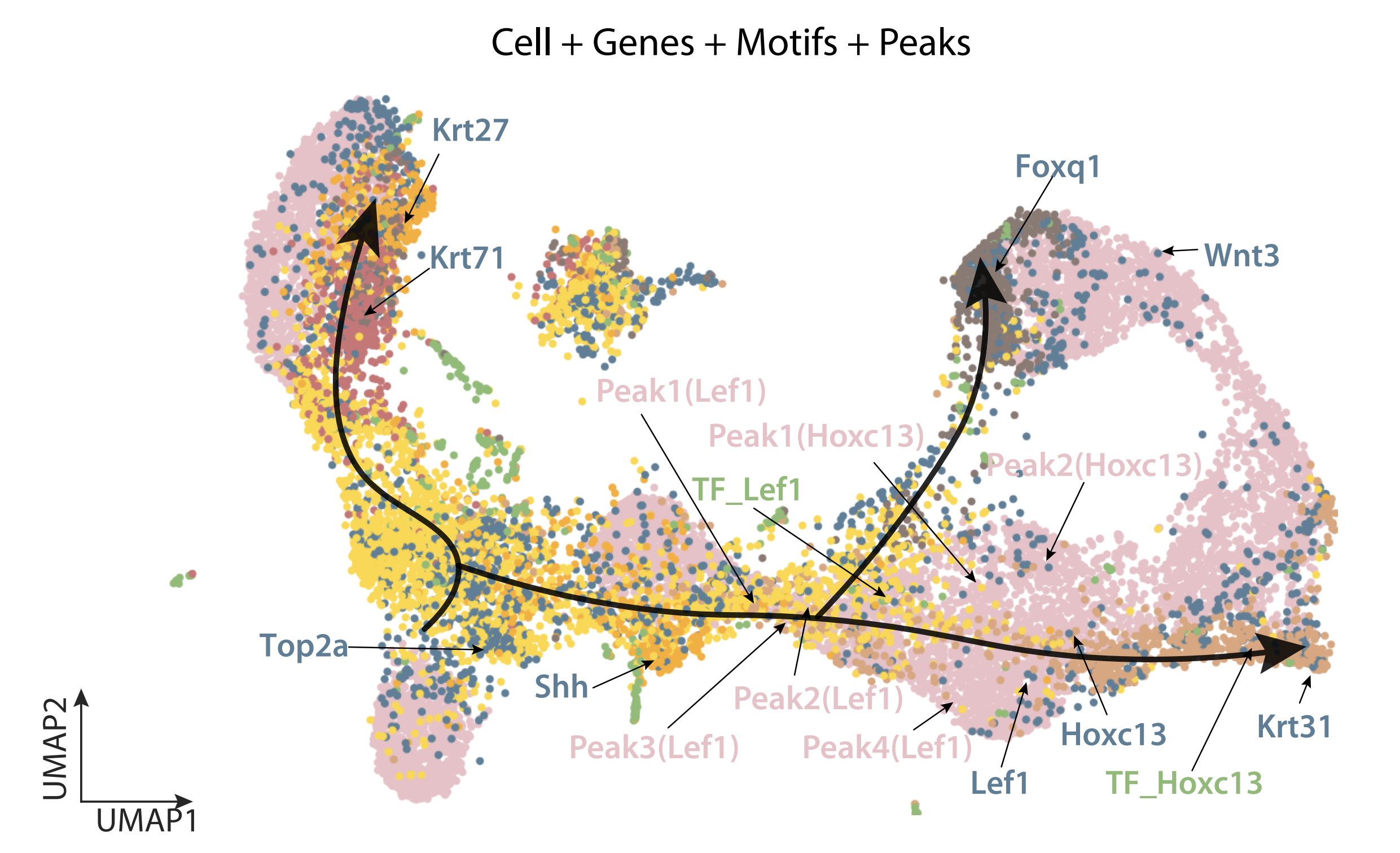
We aim to further expand to build more interpretable and generalizable framework that can work on perturbation and time series single cell data.
Optimal transport based embedding of single cell genomics data
May-Jul 2021
Based on the approach applied to Natural Language Processing to embed words, sentences and documents, I studied the how the cells and their genomic features can embedded and queried in the lower dimensional embedding.
Detecting structural variants from single cell whole genome amplification sequencing
Aug-Nov 2021
Human brain has somatic mutations that are yet uncharacterized for their action in neurological disorders including Alzheimer’s disease. I analyzed and developed structural variant detection method from single cell whole genome sequencing data amplified with primary template-directed amplification (PTA) sampled from brain in Park lab.
Prediction model from chromatin accessibility to gene expression
Feb-Jul 2020
Recent development of multiomic profiling in single cells opened a way to study the relationship between chromatin accessibility of regulatory elements and the downstream gene regulation. I analyzed fetal heart development SNARE-seq data to build and evaluate a shared prediction model from the chromatin accessibility to the gene expression in Kharchenko lab.
Single-cell expression profiling of metastatic bladder cancer
Oct-Dec 2019
Single cell profiling enables the study of heterogeneity, and can give major insigts on cancer biology including the composition of the cancer and their properties. I identified the heterogeneity in metastatic bladder cancer samples in Van Allen lab.
Characterizing the new class of super-enhancers that are shared across different cell types
Jan-Oct 2019
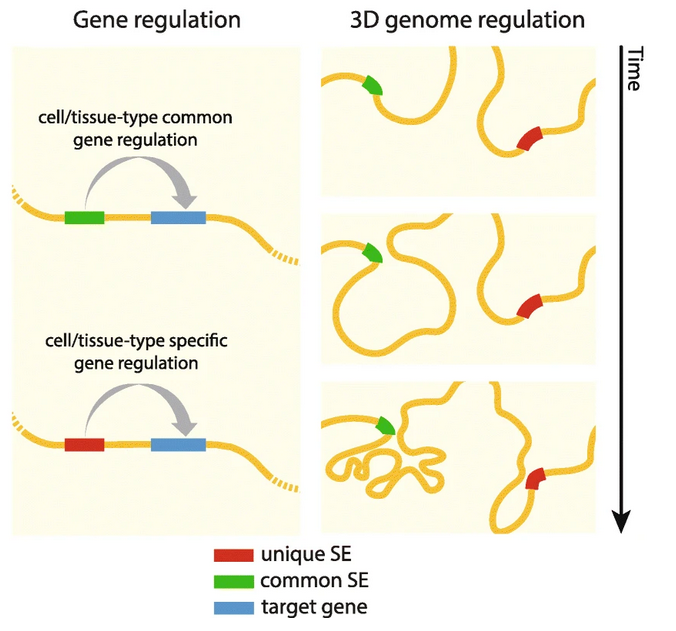 Super-enhancers are a large genomic region that contains multiple regulatory elements showing strong activation signal. Previously, they have been known to be highly cell type specific to define different cell types. In Jung lab, our team identified a group of super-enhancers that are shared across numerous cell types. Those constitutive super-enhancers are associated with the fast recovery of chromatin loops after cohesin disruption, suggesting their regulatory role in chromatin organization. This project resulted in first author publication and oral presentation in international conference.
Super-enhancers are a large genomic region that contains multiple regulatory elements showing strong activation signal. Previously, they have been known to be highly cell type specific to define different cell types. In Jung lab, our team identified a group of super-enhancers that are shared across numerous cell types. Those constitutive super-enhancers are associated with the fast recovery of chromatin loops after cohesin disruption, suggesting their regulatory role in chromatin organization. This project resulted in first author publication and oral presentation in international conference.
Network analysis of a model for effective functional residue prediction from multiple sequence alignment
Mar-Dec 2017
Advisor: Dongsup Kim